


INTRODUCTION
The newborn is prone to early brain injury caused by perinatal asphyxia, stroke, birth trauma, metabolic imbalances, genetic disorders and seizure activity. Most survivors of perinatal brain injury exhibit neurologic deficits that persist, such as cerebral palsy, mental retardation, or epilepsy.1 Treatment and care for the sequelae of early brain injury require significant resources. Even after maximal care, there is often only little improvement in the overall abilities of an individual with long-term effects on the family, healthcare system and society. Despite important progress in obstetric and neonatal care during the last decades, perinatal hypoxic injury remains one of the most important causes of neonatal brain injury and its associated adverse developmental outcome.2,3 The severity, intensity and timing of
asphyxia, as well as a selective ischemic vulnerability and the immaturity of the brain determine the extension and the degree of severity of the ensuing damage and long-term neurodevelopmental impairment.4,5
Neuropathological studies indicate that many critical neuronal groups are more vulnerable to injury in newborns (immature brain) than in adults, particularly related to enhanced density and function of excitatory amino acid receptors as well as enhanced vulnerability to attack by reactive oxygen species (ROS) and reactive nitrogen species.6 In fact, the immature brain has more blood vessels, higher water content, lower myelin, a poorly developed cortex, and a more prominent germinal matrix than the mature brain.7 These characteristics make the preterm brain more susceptible to hypoxic ischemic damage. Due to these immature brain characteristics, it is necessary to focus on the period of time following the injury-causing event, when the therapeutic strategies could be efficacious in the reduction of brain damage. This period, being normally short and varying from 2 to 6 hours, a rapid identification could facilitate the application of diverse rescue strategies.
A search for “neuroprotective” therapies that can prevent injury progression or enhance repair of the immature brain continues with the goal of improving long-term neurologic outcomes. Optimizing the therapy for early brain injury requires targeting on multiple pathways that not only prevent cell death, but also enhance cell growth, cell differentiation and integration into neural networks. In addition to neuronal damage, injury to non-neuronal cell types, such as oligodendrocytes and astrocytes should also be addressed to optimize results. The goal of non-pharmacologic and pharmacologic therapies is to target the response cascade to injury and salvage cells that would otherwise die, protect cells from becoming injured or at risk for death by increasing tolerance, repair injured cells and enhance neurogenesis. Therapies enhancing the protective and reparative processes may need to be combined and the optimal time of intervention needs to be sought for.
PATHOGENESIS OF NEONATAL BRAIN INJURY
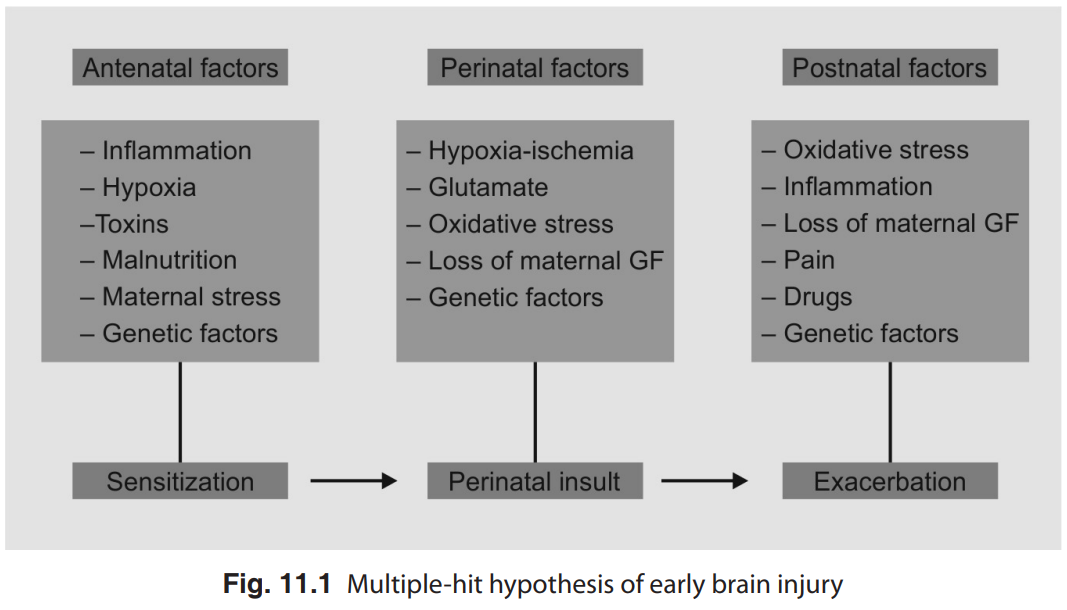
Early brain damage is thought to be mediated and modulated by multiple processes and predisposing factors. The multiple hit hypothesis has been proposed to explain this (Fig. 11.1) in which a combination of two or more environmental or genetic factors operating during the prenatal, perinatal, or postnatal period induces or modulates brain lesions in human neonates.
Brain damage following a perinatal hypoxic ischemia is an evolving process, which is comprised of two phases. A first phase consists of an early/primary energy failure, where the oxidative energy metabolism of cells decreases and it promotes necrotic death. This is followed by a second phase of cell death, a late/secondary energy failure, which occurs during reperfusion and reoxygenation several hours after the initial event and lasts for days., The pathophysiology of this late/secondary energetic failure
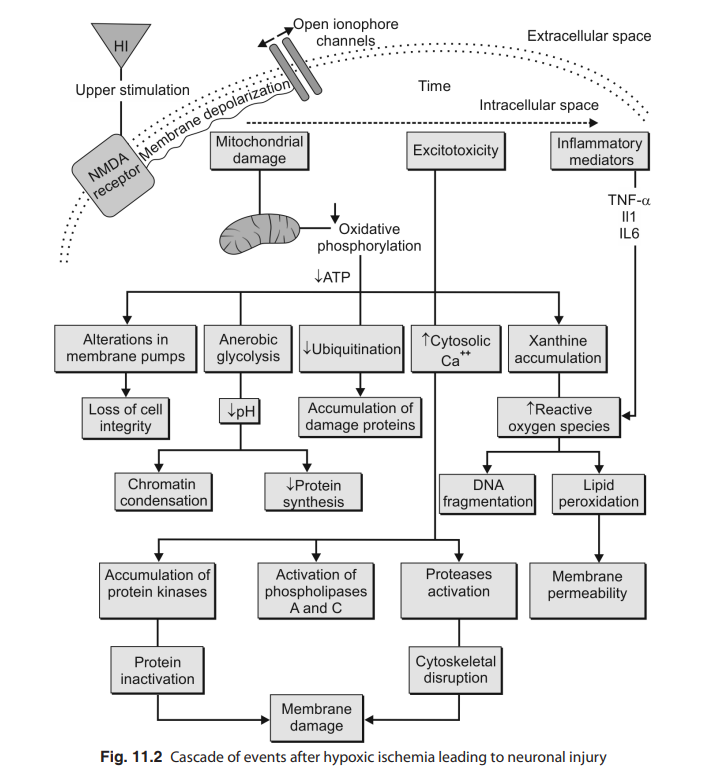
initiates a cascade of biochemical events (Fig. 11.2), which involve nitric oxide synthases activation, the production of cytotoxic free radicals, inflammation, membrane dysfunction and apoptosis, among others.7-10
The mechanism of brain injury is thought to involve a series of events that we refer to as the “excito-oxidative cascade”. Activation of excitatory glutamate receptors, especially N-Methyl-D-aspartate (NMDA) receptors, occurs very early during the initial hypoxic-ischemic insult. Then the oxidative stress associated with worsening mitochondrial dysfunction and mitochondrial failure becomes an important factor that determines whether neurons and glia survive or die by apoptosis or necrosis. This cascade probably accounts for the process of secondary energy failure in mitochondria, in which the brain’s energy supplies fall over a period of about 24 hours in babies.11-13
Glutamate-receptor-mediated Injury
The initial events to occur during a severe hypoxic-ischaemic episode associated with metabolic acidosis are depolarization of neuronal membranes and power failure of glutamate transporters on perisynaptic glia that ordinarily remove the neurotransmitter from the synaptic cleft (Figure 11.1). Hypoxia- ischaemia reduces ionic gradients across the neuronal membranes leading to membrane depolarization and neurotransmitter release. Glutamate reuptake transporters can operate anerobically in hypoxemic conditions, but become impaired when ischemia secondary to falling cardiac output restricts the delivery of glucose. Transporter failure then causes glutamate to accumulate within synapses and spill over into the brain’s extracellular space.14-17 This accumulation is consistent with notable increases in concentrations of the excitatory aminoacids glutamate and aspartate in the cerebrospinal fluid (CSF) of babies with moderate or severe encephalopathy.18,19 Studies in fetal lambs have also shown that hypoxia-ischemia causes extracellular overflow
of glutamate and other aminoacids in the cortex and basal ganglia.20 Loss of membrane potential, combined with high concentrations of glutamate, opens calcium-permeable NMDA glutamate channels and voltage-gated calcium channels, allowing calcium to move into neurons.21 Strong evidence suggests that many types of calcium conductance channels are open during sustained hypoxic-ischemic-induced depolarization; substantial non-specific transmembrane leakage also occurs.21 Calcium flooding into cells activates a cascade of events that can cause cell death. Endogenous activation of adenosine A1 receptors during severe asphyxia mediates
the initial suppression of neural activity and is an important protective mechanism.22 Pretreatment of 7-day-old rats with the NMDA channel blocker MK-801 before hypoxia-ischemia is strongly protective, but the temporal window for protection by this drug after hypoxic ischemia is less than 3 hours,20 suggesting that the cascade downstream of NMDA receptors quickly becomes irreversible, self-perpetuating and unresponsive to channel blockade. Magnesium is neuroprotective against the damage caused by Hypoxic ischemic encephalopathy (HIE) in neonatal rodent models, probably due to its ability to block NMDA-receptor channels.23 a-amino-3-hydroxyl-5 methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors are also activated by excess glutamate and probably contribute to seizures during hypoxic-ischemic encephalopathy. The prominence of NMDA-mediated injury in the immature brain is related to the fact that NMDA receptors are functionally upregulated in the perinatal period because of their role in activity-dependent neuronal plasticity.24 Immature NMDA channels open more easily and stay open longer than do adult channels, and the voltage- dependent magnesium block, that is normally present in adult channels at resting membrane potentials, is more easily relieved in the perinatal period. Increased expression of NMDA receptor subtype 2B (NR2B) subunits on NMDA receptors is thought to be responsible for their greater excitability and longer open time during the neonatal period.
IDENTIFYING BABIES AT RISK
A variety of clinical predictors have been used to identify those at risk of hypoxic brain injury. These include low Apgar scores, cord blood or early arterial acidosis, and seizures or the presence of encephalopathy on examination. To identify encephalopathy Electroencephalography (EEG) and cerebral function monitoring using amplitude-integrated EEG (aEEG) are being increasingly used. Brain imaging with magnetic resonance imagimg (MRI), including newer techniques such as spectroscopy (MRS), diffusion weighted (DWI) and diffusion tensor imaging (DTI), and volumetric analysis provides the most accurate assessment of injury. These techniques allow the determination of the severity and evolution of brain injury, with specific injury patterns being associated with poor outcomes such as loss of grey/white matter differentiation, watershed injury, and thalamic or basal ganglia injury. However, the early and sequential brain imaging in the neonates is often not possible because of scanner availability and difficulty in transporting these critically ill patients. Biomarkers of oxidative stress and inflammation, or indicators of other organ injuries are being looked into but have till now being of equivocal value in identifying early brain injury. Given all the available evidence, a combination of neurologic examination and seizue occurrence provides the best estimate of babies who may be at risk for brain injury.25
NEUROPROTECTIVE THERAPIES/STRATEGIES
Hypothermia
Hypothermia has been used successfully since the 1950s as a protective measure to allow the babies with complex congenital heart disease to undergo circulatory arrest for corrective surgery, but its use in neonatological settings was impeded by the data showing that it increased the death rate in premature infants. However, interest in this technique was renewed with the publication of data on the pathogenesis of HIE reviewed above, especially that which showed the importance of delayed secondary energy failure and the excito-oxidative cascade in neurological damage. Experiments that showed efficacy and safety in clinically relevant large animal models were especially important in bringing this technique to the clinic.
Mechanism of Hypothermic Neuroprotection
Although our understanding of the mechanisms for the neuroprotective effect of hypothermia remains incomplete, experimental studies have shown that it inhibits many steps in the excito-oxidative cascade including secondary energy failure, increases in brain lactic acid, glutamate, and nitric oxide concentrations, and epileptic activity. Protective effects have also been associated with inhibition of protease and calpain activation, loss of mitochondrial membrane potential and mitochondrial failure, free radical damage, lipid peroxidation and inflammation. Experiments in which the glutamate agonist NMDA was injected directly into the brains of 7-day- old rat pups to produce excitotoxic lesions showed that injury was directly proportional to temperature over a range of 25–40°C. This suggests that hypothermia can reduce injury triggered by NMDA-receptor activation alone without hypoxia-ischemia. Ikonomidou and colleagues also showed that hypothermia enhances the protective effect of the noncompetitive NMDA channel antagonist MK-801 in a model of hypoxic-ischemic brain injury in rat pups. Brief periods of moderate hypothermia to 32°C after injury have been shown to delay cell death in 7-day-old rat pups by as much as a week, after which brain atrophy occurs.26-28 These results suggest that moderate hypothermia can delay the programmed cell death, independent of its role in other aspects of metabolism, and might be able to extend the therapeutic window for other interventions.
Clinical Trials
Among the many human newborn trials of therapeutic hypothermia (Table 11.1), the publication of three randomized controlled trials of moderate hypothermia or conventional treatment for HIE in term infants, published from 2005 to 2009, is generally thought to have been a milestone in this area of neonatology. The first study, the CoolCap trial29 by Gluckman and colleagues, included 234 term babies less than 6 hours old with evidence of asphyxia at birth (Apgar <5 at 10 min, or severe acidosis) and background abnormality

on a EEG. The group was randomized to treatment with a selective head cooling device to maintain rectal temperature at 34.5°C for 72 hours or to conventional care. The primary outcome was death or severe disability at 18 months when a neurological and developmental assessment was done on a total of 218 children who received cooling or conventional treatment. There were no significant differences between groups for the primary outcome for all children but outcome was significantly better for hypothermia when the pre-stratified group of children with the most severe encephalopathy on an EEG were excluded. This suggested that babies with severe encephalopathy were unresponsive to cooling.
The second study was the National Institutes of Child Health and Human Development (NICHD) whole-body hypothermia for neonates with HIE by Shankaran and colleagues.30 They enrolled 208 term infants less than 6 hours old with evidence of HIE associated with asphyxia who were randomized to total body cooling to 33.5°C for 72 hours or conventional care. Follow-up at 18 months of age in 205 of 208 infants showed that cooling significantly reduced the primary endpoint of death or moderate or severe disability from 62% in controls to 44% in cooled infants.
Thethirdtrialwasthetotalbodyhypothermiafor Neonatal Encephalopathy Trial (TOBY) by Azzopardi and colleagues.31 This study enrolled 325 infants with entry criteria that were identical to those of the CoolCap trial, and infants were randomized to cooling at 33.5°C for 72 hours or conventional care. Infants in the cooled group were significantly more likely to survive without neurological abnormality (44% cooled vs 28% non-cooled), and had lower rates of cerebral palsy (33% cooled vs 48% noncooled) and higher developmental scores than non-cooled infants.
Studies of therapeutic hypothermia in human neonates show a reduction in mortality and long-term neurodevelopmental disability at 12–24 months of age, with the most benefit seen in moderately encephalopathic infants.
Sustained protection does depend on the dose of hypothermia, with maximum benefit obtained with cooling to 33–34°C, as well as on limited delay to treatment initiation. Mild hypothermia to this level appears to be well tolerated without serious adverse effects, if initiated within the first 6 hours of life.
Three meta-analyses of clinical trials of moderate hypothermia for HIE have been published since publication of the TOBY trial, one by Edwards and colleagues,32 another by Shah,33 and a recent Cochrane meta analysis by Jacobs and colleagues.34 Edwards and colleagues analyzed the TOBY, CoolCap, and the NICHD trials together with seven other trials and concluded that cooling reduced the risk of death or disability at 18 months with a number needed to treat of nine with little risk of adverse effects and Shah’s analysis was generally consistent with this assessment. Jacobs and colleagues in their Cochrane systemic review of 11 trials (N=1505 infants) concluded that therapeutic hypothermia is beneficial in term and late preterm newborns with hypoxic ischemic encephalopathy. Cooling reduces mortality without increasing major disability in survivors. The benefits of cooling on survival and neurodevelopment outweigh the short-term adverse effects. Hypothermia should be instituted in term and late preterm infants with moderate-to-severe hypoxic ischaemic encephalopathy if identified before six hours of age. Further trials to determine the appropriate techniques of cooling, including refinement of patient selection, duration of cooling and method of providing therapeutic hypothermia, will refine our understanding of this intervention.
Therapeutic hypothermia has now become standard of care for neonatal HIE brain injury. Currently, there are two main ways to provide cooling therapy (Figure 11.3): cooling of the head with or without mild systemic cooling, or cooling the body alone. Selective head cooling with only minimal cooling of the body is achieved using a specialized cooling cap applied to the infant’s head with circulating cooled water; however, some concern remains that establishing the actual brain temperature, especially of deep brain

structures, is not currently possible. In contrast, whole body cooling, achieved by use of cold packs or a servo regulated cooling blanket, assumes that core temperature approximates brain temperature, which is measured by rectal probe. Whichever method is used, cooling is maintained for 72 hours at 33°C to 34°C. Selection of infants for cooling may be challenging, however, it needs to be accomplished within 6 hours of birth and preferably as soon after injury as possible. Specifically, it is important to exclude other possible causes of encephalopathy, such as metabolic disease, infection, drug exposure, central nervous system malformations and neonatal stroke in order to not expose an infant to an unnecessary treatment. Whilst various selection methods are used by individual centers, generally the following criteria need to be met. The infant must be greater than 36 weeks gestation, aged less than six hours, in moderate or severe encephalopathy as per Sarnath staging, with at least one of the following:
- Apgar score of <5 at 10 minutes or continued need for assisted ventilation for at least 10 minutes
- Any acute perinatal event that may result in HIE (e.g. placental abruption, cord prolapse, severe fetal heart rate abnormality)
- Cord pH <7.0 or BE > 16 mmol/L
- Blood gas within 60 minutes of birth with pH <7.0 or BE > 16 mmol/L.
An amplified electroencephalogram (aEEG) may be helpful for detecting abnormal brain activity and/or seizures; however it is important not to delay cooling in order to obtain an aEEG if the above criteria are met.
Growth Factors
Animal studies show that milder forms of neurologic injury leads to a preconditioning of neurological responses to subsequent more severe injuries with the protective effects possibly lasting for weeks. Hypoxia- inducible factor 1a (HIF-1a) activation is a key modulator of the protection against subsequent hypoxic ischemic injury that is induced by hypoxic preconditioning. HIF-1a produces a variety of downstream targets that are neuroprotective, including insulin-like growth factor-1 (IGF-1), vascular endothelial growth factor (VEGF), and erythropoietin (EPO).35
Erythropoietin
Research has recently indicated that erythropoietin a 34-kDa glycoprotein traditionally used to enhance erythropoiesis, may also provide neuroprotection to infants with HIE. EPO receptors (EPO) are present in the developing human embryo, and higher levels of EPO in cerebral spinal fluid have been correlated with improved neurodevelopmental outcomes. EPO may benefit infants with HIE through protection from neuronal apoptosis, neural regeneration, decreased inflammation, and decreased susceptibility to glutamate toxicity. Preterm infants treated with EPO for promotion of erythropoiesis have demonstrated improved neurodevelopmental outcomes.
Term infants with HIE treated with EPO show decreased seizure activity, improved EEG results, and enhanced neurologic outcome.36,37 Although additional clinical trials are needed, EPO appears to be effective in the treatment of infants with HIE if administered within 48 hours after delivery.
In humans, EPO is safely used for treatment of anemia in premature infants. EPO for neuroprotection is given in much higher doses (1000-5000 U/ kg/dose) than for anemia, to enable crossing of the blood-brain barrier, with unknown pharmacokinetics in humans. Recently, extremely low birth weight infants tolerated doses between 500 and 2500 U/kg/dose.38 Concern also exists regarding potential adverse reactions associated with EPO. Although significant side effects including hypertension, clotting abnormalities, seizures, and polycythemia have not been seen in neonates, they have been reported in the adult population. In addition, one study reported an increased incidence of retinopathy of prematurity in premature infants treated with epogen, which is of particular concern if high doses are to be administered. Additional research is required to more adequately assess whether EPO is an effective therapy for HIE and to answer questions regarding optimal dosage, timing, and length of treatment.
Vascular Endothelial Growth Factor (VEGF)
VEGF is a regulator of angiogenesis that is also involved in neuronal cell proliferation and migration. Following exposure to hypoxia, there is increased neuronal and glial expression of VEGF-A, directing vascularization and stimulating proliferation of neuronal and non neuronal cell types. VEGF also has chemotactic effects on neurogenic zones in the brain, increasing migration of stem cells during anoxia. In adult ischemia models, VEGF administration has shown improved outcomes, and studies are ongoing to assess its utility in neonatal neuronal injury situations.39
Other trophic factors, like insulin-like growth factor (IGF-1) and brain derived neurotrophic factor (BDNF) have also shown promise, but given their role in normal neurodevelopment, the effects of treatment are not known.
Stem Cell Therapy
Stem cells have the ability to differentiate into a variety of cell types in the central nervous system. Stem cell transplantation may minimize the effect of early brain injury by replacing damaged cells, promoting cell regeneration, inhibiting inflammation, and releasing trophic factors that heal and improve cell survival. In adult animal models of brain injury implanted stem cells integrate into injured tissue, decreasing volume loss and improving behavioral outcomes. In neonatal models, intraventricular implantation of neonatal stem cells (NSC) after HIE results in their migration to injured areas, and differentiation into neurons, astrocytes, oligodendrocytes, and undifferentiated progenitors. These cells promote regeneration, angiogenesis and neuronal cell survival in both rodent and primate models, and non-neuronal progeny inhibit inflammation and scar formation.40,41
Efficacy of stem cell transplantation, however, appears dependent on timing of implantation, and this therapeutic window is presently unknown. So, although stem cell therapy for early brain injury appears promising, a significant amount of research is required before it can be considered for use in neonates.
Antioxidants
Oxidative stress is an important component of early injury to the neonatal brain. Antioxidant defenses, such as superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase, and compounds, such as vitamins A, C, E, beta carotene, glutathione and ubiquinones scavenge free radicals under normal conditions. Damage occurs when there is an imbalance between their generation and uptake. Following HI, there is an increase in superoxide and hydroxyl radical production and rapid depletion of antioxidant stores, which leads to cell membrane damage, excitotoxic energy depletion, cytosolic calcium accumulation, and activation of proapoptotic genes that cause damage to cellular components and result in cell death. The neonatal brain has a high rate of oxygen consumption and low concentration of antioxidants, making it susceptible to damage. An inducible isoform of nitric oxide synthase (iNOS) produces nitric oxide (NO) in response to cellular stress, which initiates neuronal damage when converted to secondary reactive nitrogen species that facilitate nitration and nitrosylation reactions. Early endothelial NO is protective by maintaining blood flow, but early neuronal NO and late inducible NO promote cell death. Brain iNOS is induced in multiple cell types during upregulation of the pro-inflammatory pathway after brain injury, modifying binding to NMDA receptors and enhancing excitotoxicity. Selective inhibition of nNOS or iNOS has shown potential as a neuroprotective strategy. There have been few studies in human newborns examining cerebral NO production. Cerebrospinal fluid (CSF) NO levels increase with severity of HI encephalopathy at 24 to 72 hours after asphyxia, with increased NO and nitrotyrosine levels in the spinal cord as well.42 Initial results in premature infants treated with inhaled NO for prevention of bronchopulmonary dysplasia show reductions in ultrasound-diagnosed brain injury and improvements in neurodevelopmental outcomes at 2 years of age. Several other antioxidant strategies that either block free radical (FR) production or increase antioxidant defenses are being studied. Melatonin is an indoleamine that is formed in higher quantities in adults and functions as a direct scavenger of ROS and NO. It has been found to provide long lasting neuroprotection in experimental HI and focal cerebral ischemic injury and human neonates treated with melatonin were also found to have decreased pro-inflammatory cytokines.
Allopurinol
Allopurinol is an antioxidant that inhibits formation of the free radicals that play a significant role in the cellular damage associated with HIE. Early
allopurinol in asphyxiated infants improved long term neurodevelopmental outcomes.43 Administration of allopurinol to mothers whose pregnancies are complicated by fetal hypoxia, however, has been associated with improved cord gases, and a large multicenter randomized control trial is currently underway to examine the effect of prenatal administration of allopurinol to fetuses at risk for HIE.44,45 Because it appears early treatment with allopurinol is optimal, prenatal treatment with placental transfer to the fetus may be the most viable option. Thus, additional research is needed to determine the effectiveness of treating infants with this innovative therapy.
Deferoxamine
Deferoxamine (DFO) is an iron chelator that decreases FR production by binding with iron and decreasing the production of OH that occurs via the Fenton reaction, while also stabilizing HIF-1a to produce its downstream products VEGF and EPO. DFO is protective during exposure to H2O2 or excitotoxicity in vitro, and in animal models of HI and transient ischemic stroke in vivo.46 N-acetylcysteine (NAC) is a glutathione precursor and FR scavenger that attenuates lipopolysaccharide-induced white matter injury in newborn rats47, but results for other antioxidant compounds, such as vitamin E, have been inconclusive.
Anti-excitotoxic/Glutamergic Inhibitors
Excitotoxicity refers to excessive glutamatergic activation that leads to cell injury and death. Glutamate accumulates in the brain after neuronal injury from a variety of causes, including vesicular release and reversal of glutamate transporters. Glutamatergic receptors include N-methyl-D- aspartate (NMDA), alpha-3-amino-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and kainate. NMDA receptor activation, while important for synaptic plasticity, can increase intracellular calcium and pro-apoptotic pathways via caspase-3 activation if overactivated. Excitotoxicity has long been known to play a part in the progression of brain injury, and differences in receptor expression contribute to the vulnerability of the developing brain. There has long been a search for agents that decrease brain injury by decreasing excitotoxicity.
Dizocilipine (MK801)
It is a non-competitive NMDA receptor antagonist that has been studied in humans. The major drawbacks are that it is poorly tolerated and has also been shown to increase apoptosis and decrease neuronal migration in animal models.48
Memantine
It is a low affinity non-competitive NMDA receptor antagonist that is well tolerated in adults for Alzheimer’s-type dementia. Post-HI treatment with
memantine attenuates acute white matter injury in rat models, resulting in long term histological improvement in vivo and restoring neuronal migration in vitro.49
Topiramate
It is an AMPA kainate receptor antagonist that is an FDA-approved anti- epileptic for patients greater than 2 years of age. It has been shown to protect newborn rodents from excitotoxic brain lesions [153], reducing brain damage and cognitive impairment when administered within two hours of the injury.50 An intravenous (IV) preparation of topiramate does not yet exist for human use, but this treatment shows potential as a therapy for early newborn seizure and injury.
Magnesium Sulfate
Magnesium sulfate (MgSO4), traditionally used for the treatment of pregnancy-induced hypertension and prevention of preterm labor, is being investigated as a neuroprotective agent for infants with HIE. Magnesium sulfate is an N-methyl-D-aspartate receptor antagonist. N-methyl-D- aspartate is a receptor for glutamate, an amino acid important in cell proliferation, differential, and survival in the developing brain. With HIE, glutamate accumulates in abnormally high levels, resulting in neuronal cell injury and death. Conflicting data exist regarding the effectiveness of MgSO4
as a neuroprotective agent.51 Prenatal administration of MgSO4 to mothers at risk for preterm delivery is associated with reduced incidence of cerebral palsy at 3 years and improved neurodevelopmental outcomes.52 However,
these benefits are not universal. When MgSO4 was administered postnatally to term infants with HIE, there was no improvement in their amplitude- integrated EEG, and when administered in large doses, MgSO4 can cause profound hypotension.
Anti-inflammatory Therapy
Maternal infection is a known risk factor for white matter damage and poor outcomes, such as cerebral palsy. The inflammatory response and cytokine production that accompanies infection may play a large role in cell damage and loss. Local microglia are activated early and produce pro-inflammatory cytokines, such as TNF-a, IL-1b and IL-6, as well as glutamate, free radicals, and nitric oxide. Systemic administration of these cytokines increases excitotoxic lesions, while therapies that block microglial activation and cytokine release, protect the brain from excitotoxic damage.
Minocycline
It is a tetracycline derivative that crosses the blood-brain barrier and has anti-inflammatory effects, including decreasing microglial activation and caspase-3 expression, lipid peroxidation, and other pro-inflammatory activity, while increasing antiapoptotic gene expression. Minocycline has
shown promise in a number of animal models of neurodegenerative or ischemic disease. In the neonatal brain, minocycline appears to decrease tissue damage and caspase-3 activation in rodents when given immediately before or after injury, but results are inconsistent.53,54
Cell Death Inhibitors
Apoptosis is a critical component of normal brain development. While necrosis plays a major role in early neuronal death in both the immature and mature brain following injury, a spectrum of cell death that includes apoptosis occurs within the first 24 hours following neonatal HIE, and may result in heterogenous responses to antiapoptotic therapies. It is also probable that apoptosis and cleavage and activation of caspase-3 are responsible for more of the cell death that occurs in delayed phases of injury and neurodegeneration. Specific and non-specific inhibition of caspases or cysteine proteases, which are highly activated after HIE, have been attempted with some success.55 For example, calpain or caspase-3 inhibitors, such as MDL 28710
and M826 protect neonatal rats after HIE.
Pretreatment with the hormone 17b-estradiol is neuroprotective in immature rats, and appears to work through both anti-apoptotic and free radical scavenging pathways. In addition, the nuclear enzyme poly (ADP-ribose) polymerase (PARP) is activated during stress and enables DNA repair; however, the PARP-1 isoform also contributes to ischemic neuronal injury by depleting energy stores and activating microglia, leading to cell death. PARP-1 is more abundant in the immature brain, and its blockade protects against excitotoxicity and ischemic injury.
Combination Therapy
In the search for an optimal neuroprotective strategy, it is probable that a combination of treatments may prove the most beneficial. Single therapy that attacks any of the aforementioned injury pathways, often results in only mild improvement. Hypothermia has become the standard of care in many institutions since showing benefit in moderately encephalopathic newborns; however, it does not completely protect or repair an injured brain, and benefits may not necessarily be long lasting. Hypothermia may be used to prolong the therapeutic window, allowing time for additional treatments to be more effective. The combination of a prenatal therapy (e.g. erythropoietin or MgSO4) with hypothermia may also prove successful. Combinatorial therapy may provide more long lasting neuroprotection, salvaging the brain from severe injury and deficits while also enhancing repair and regeneration, hopefully providing additive, if not synergistic, protection.
Xenon is approved for use as a general anesthetic in Europe and has shown promise as a neuroprotective agent. It is an NMDA antagonist, preventing progression of excitotoxic damage. Combination of xenon and hypothermia initiated 4 hours after neonatal HI provided synergistic histological and functionalprotectionwhenevaluatedat 30 daysafterinjury.56 N-acetylcysteine
(NAC) is a medication approved for neonates that is a scavenger of oxygen radicals and restores intracellular glutathione levels, attenuating reperfusion injury and decreasing inflammation and NO production in adult models of stroke.57,58 Adding NAC therapy to systemic hypothermia reduced brain volume loss at both 2 and 4 in rodent models of hypoxic injury. Inhibition of inflammation with MK-801 has also been effective when combined with hypothermia in neonatal rats post HI injury.59 In P7 rats who underwent HI followed by early topiramate and delayed hypothermia, improved short- term histology and function was seen.60 The inhibition of inflammation may provide a window for protection if hypothermia is delayed, which is possible given difficulty in initiation of cooling if infants are born at an outside hospital or transport is delayed. More extensive studies in human neonates are necessary before the potentially promising combination therapies become standard recommendations.
REFERENCES
- Dilenge ME, Majnemer A, Shevell MI. Long-term developmental outcome of asphyxiated term J Child Neurol 2001; 16:781–792.
- Glass, HC; Ferriero, DM. Treatment of hypoxic-ischemic encephalopathy in newborns. Curr Options Neurol 2007;9:414–423.
- Carl G, Reiger , Evans N. One-year neurodevelopmental outcome after moderate newborn hypoxic ischemic encephalopathy. J Paediatr Child Health 2004;40: 217–220.
- Chau V, Clement JF, Robitaille Y, D’Anjou G, Vanasse Congenital axonal neuropathy and encephalopathy. Pediatr Neurol 2008;38:261–266.
- Miller SP, Ramaswamy V, Michelson D, et Patterns of brain injury in term neonatal encephalopathy. J Pediatr 2005;146:453–460.
- Yager JY, Thornhill The effect of age on susceptibility to hypoxic-ischemic brain damage. Neurosci Biobehav Rev 1997;21:167–174.
- Du Plessis J, Volpe JJ. Perinatal brain injury in the preterm and term newborn. Curr Opin Neurol 2002; 15: 151–157.
- Walton M, Connor B, et Neuronal death and survival in two models of hypoxic- ischemic brain damage. Brain Res Rev 1999; 29:137–168.
- Ferriero Neonatal brain injury. N Engl J Med 2004;351:1985–1995.
- Wyatt JS, Edwards AD, Azzopardi D, Reynolds Magnetic resonance and near infrared spectroscopy for investigation of perinatal hypoxic-ischemic brain injury. Arc Dis Child 1989; 64:953–963.
- Yager, JY Brucklacher, RM, Vannucci, RC. Cerebral energy metabolism during hypoxia-ischemia and early recovery in immature rats. Am J Physiol 1992,262: 672–677.
- Johnston Excitotoxicity in perinatal brain injury. Brain Pathol 2005;15:234–240.
- Taylor DL, Edwards AD, Mehmet Oxidative metabolism, apoptosis and perinatal brain injury. Brain Pathol 1999;9:93–117.
- Silverstein FS, Buchanan K, Johnston Perinatal hypoxia-ischemia disrupts striatal high-affinity [3H]glutamate uptake into synaptosomes. J Neurochem 1986; 47: 1614–1619.
- Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS. Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic J Cereb Blood Flow Metab 2006;26:1285–1297.
- Hagberg H, Thornberg E, Blennow M, et Excitatory amino acids in the cerebrospinal fl uid of asphyxiated infants: relationship to hypoxic-ischemic encephalopathy. Acta Paediatr 1993;82:925–929.
- Riikonen RS, Kero PO, Simell Excitatory amino acids in cerebrospinal fluid in neonatal asphyxia. Pediatr Neurol 1992;8:37–40.
- Hagberg H, Andersson P, Kjellmer I, Thiringer K, Thordstein Extracellular overflow of glutamate, aspartate, GABA and taurine in the cortex and basal ganglia of fetal lambs during hypoxia-ischemia. Neurosci Lett 1987;78:311–317.
- Cross JL, Meloni BP, Bakker AJ, Lee S, Knuckey Modes of calcium entry and homeostasis following cerebral ischemia. Stroke Res Treat 2010;133:798–802.
- Takahashi T, Otsuguro K, Ohta T, Ito Adenosine and inosine release during hypoxia in the isolated spinal cord of neonatal rats. Br J Pharmacol 2010;161:1806–1816.
- Cetinkaya M, Alkan T, Ozyener F, Kafa IM, Kurt MA, Koksal Possible neuroprotective effects of magnesium sulfate and melatonin as both pre-and post-treatment in a neonatal hypoxic-ischemic rat model. Neonatology 2010;99:302–310.
- Jensen The role of glutamate receptor maturation in perinatal seizures and brain injury. Int J Dev Neurosci 2002;20:339–347.
- McDonald JW, Johnston Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Rev 1990;15: 41–70.
- Monyer H, Brunashev N, Laurie Development and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1993;12:529–540.
- Miller SP, Latal B, Clark H, et Clinical signs predict 30-month neurodevelopmental outcome after neonatal encephalopathy. Am J Obstet Gynecol 2004;190:93–99.
- Thoresen M, Penrice J, Lorek A, et Mild hypothermia after severe transient hypoxia- ischemia ameliorates delayed cerebral energy failure in the newborn piglet. Pediatr Res 1995;37:667–670.
- Drury PP, Bennet L, Gunn Mechanisms of hypothermic neuroprotection. Semin Fetal Neonatal Med 2010;15:287–292.
- Ikonomidou C, Mosinger JL, Olney Hypothermia enhances protective effect of MK-801 against hypoxic/ischemic brain damage in infant rats. Brain Res 1989;487: 184–187.
- Gluckman PD, Wyatt JS, Azzopardi D, et Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicenter randomized trial. Lancet 2005;365:663–670.
- Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole body hypothermia for neonates with hypoxic-ischemic N Engl J Med 2005;353:1574–84.
- Azzopardi DV, Strohm B, Edwards AD, et Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med 2009;361:1349–1158.
- Edwards AD, Brocklehurst P, Gunn AJ, et Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 2010;340–363.
- Shah Hypothermia: a systematic review and meta-analysis of clinical trials. Semin Fetal Neonatal Med 2010;15:238–246.
- Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev 2013;(1):CD003311.
- Ran R, Xu H, Lu A, Bernaudin M, Sharp Hypoxia preconditioning in the brain. Dev Neurosci 2005;27:87–92.
- McPherson RJ, Juul SE. Erythropoietin for infants with hypoxic-ischemic encephalopathy. Curr Opin Pediatr 2010;22:139–145.
- Zhu C, Kang W, Xu Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics 2009;124:e218–226.
- Juul SE, McPherson RJ, Bauer LA, Ledbetter KJ, Gleason CA, Maycock A Phase I/II Trial of High Dose Erythropoietin in Extremely Low Birth Weight Infants: Pharmacokinetics and Safety. Pediatrics 2008;122:504–510.
- Zhang ZG, Zhang L, Jiang Q, et VEGF enhances angiogenesis and promotes blood- brain barrier leakage in the ischemic brain. J Clin Inves 2000;106:829–838.
- Park KI, Himes BT, Stieg PE, Tessler A, Fischer I, Snyder Neural stem cells may be uniquely suited for combined gene therapy and cell replacement: Evidence from engraftment of Neurotrophin-3-expressing stem cells in hypoxic-ischemic brain injury. Exp Neurol 2006;199:179–190.
- Imitola J, Raddassi K, et Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci U S A 2004;101:18117–18122.
- Groenendaal F, Vles J, Lammers H, De Vente J, Smit D, Nikkels Nitrotyrosine in human neonatal spinal cord after perinatal asphyxia. Neonatology 2008;93:1–6.
- Kaandorp JJ, van Bel F, et Long-term neuroprotective effects of allopurinol after moderate perinatal asphyxia: follow-up of two randomized controlled trials. Arch Dis Child Fetal Neonatal Ed 2012;97:F162-166.
- Torrance HL, Benders MJ, Derks JB, et Maternal allopurinol during fetal hypoxia lowers cord blood levels of the brain injury marker s-100b. Pediatrics 2009;124: 350–357.
- Kaandorp JJ, Benders MJ, Rademaker CM, et Maternal allopurinaol for reduction of birth asphyxia induced brain damage (allo-trial): a randomized double blind placebo controlled multicenter study. BMC Pregnancy Childbirth. 2010;10:72–81.
- Sarco DP, Becker J, Palmer C, Sheldon RA, Ferriero The neuroprotective effect of deferoxamine in the hypoxic-ischemic immature mouse brain. Neurosci Let 2000;12: 282.
- Paintlia MK, Paintlia AS, Barbosa E, Singh I, Singh N-acetylcysteine prevents endotoxininduceddegenerationofoligodendrocyteprogenitorsandhypomyelination in developing rat brain. J Neurosci Res 2004;78:347–361.
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 2002;1:383–386.
- Volbracht C, van Beek J, Zhu C, Blomgren K, Leist Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci 2006;23:2611–2622.
- Noh MR, Kim SK, Sun Neuroprotective effect of topiramate on hypoxic ischemic brain injury in neonatal rats. Exp Neurol 2006;201:470–478.
- Khashaba MT, Shouman BO, Shaltout AA, et al. Excitatory amino acids and magnesium sulfate in neonatal Brain Dev 2006;28:375–379.
- Crowther CA, Hiller JE, Doyle LW, Haslam Effect of magnesium sulfate given for neuroprotection before preterm birth: a randomized controlled trial. JAMA 2003;26: 2669–2676.
- Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic Ann Neurol 2002;52:54– 61.
- Fox C, Dingman A, Derugin N, et Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab 2005;25:1138–1149.
- Han BH, Xu D, Choi J, et Selective, reversible caspase-3 inhibitor is neuroprotective and reveals distinct pathways of cell death after neonatal hypoxic-ischemic brain injury. J Biol Chem 2002;277:30128–30136.
- Ma D, Hossain M, Chow A, et al. Xenon and hypothermia combine to provide neuroprotection from neonatal Ann Neurol 2005;58:182–193.
- Khan M, Sekhon B, Jatana M, et al. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental J Neurosci Res 2004; 76:519–527.
- Sekhon B, Sekhon C, Khan M, Patel SJ, Singh I, Singh N-Acetyl cysteine protects against injury in a rat model of focal cerebral ischemia. Brain Res 2003; 971:1–8.
- Alkan T, Kahveci N, Buyukuysal L, Korfali E, Ozluk Neuroprotective effects of MK 801 and hypothermia used alone and in combination in hypoxic-ischemic brain injury in neonatal rats. Arch Physiol Biochem 2001; 109:135–144.
Liu Y, Barks JD, Xu G, Silverstein FS. Topiramate extends the therapeutic window for hypothermia mediated neuroprotection after stroke in neonatal rats. Stroke 2004;1460–1465.










